为什么氯气与氢气的反应需要镁条引发?
假期刷到初中的一个实验的视频,氯气和氢气在光照下化合。想起当时很不能理解明明室内开了灯,但是不能发生反应,非要镁条点一下才行。曾经问老师得到的解释是灯光强度不够,要Mg点燃的时候那个晃眼的强度才行。现在掌握的知识多了些,忽然有兴致随便算算玩玩。
首先分析一下。反应的关键是氯氯键需要被光打断,解离成氯自由基。这个过程涉及到氯气的激发和断键,应该是吸收到S1然后过MECI断键。既然涉及到吸收,那首先需要了解一下自然光的光谱。日常生活里,常见的光基本上有两种:
i. LED电灯。为了避免紫外辐射对人眼和材料的伤害,商业LED灯通常会滤掉紫外成分。找了些发射光谱,看到它的发射大概在450-700 nm之间,其中450 nm有一个尖锐的峰,再往蓝走强度锐减,400 nm以前基本都没有了。
ii. 日光。取
ASTM G-1731的数据,太阳光到达地面的光谱(节选)大概长这样:
虽然紫外段的强度低些,但320 nm以上的光算是都有。
氯氯键的经典键能在242.6 kJ/mol,对应波长495 nm。那似乎确实这两种光都可以引发反应。但是笔者还是觉得有点奇怪,为什么自由基反应大多都要强调日光,而Mg又凭什么可以?
氯气的吸收
体系很小,CCSD优化一下键长看看:
1
2
3
4
5
6
7
8
9
10
%nproc=32
%mem=96GB
%chk=cl2.chk
# CCSD/def2tzvp opt freq=num
bane
0 1
Cl 0.0 0.0 0.0
Cl 0.0 0.0 2.00
换了三个基组测试:
| 基组 | 键长 |
|---|---|
| def2-tzvp | 2.00277 |
| cc-pvtz | 2.00943 |
| def2-qzvp | 1.98850 |
查到实验值是1.988,qzvp的结果还挺接近的,后面用这个键长当作平衡键长值了。
接着用NEVPT2算激发能。算简单的结构,直接交给MOKIT其实挺稳的,事后检查基本都没啥问题
1
2
3
4
5
6
7
8
9
%nproc=32
%mem=96GB
# NEVPT2/cc-pvtz
mokit{Nstates=2}
0 1
Cl 0.000000 0.000000 0.994248
Cl 0.000000 0.000000 -0.994248
这里MOKIT取的应该是CIS的NO作为初猜
1
2
3
4
CIS energies from Gaussian:
Excited State 1: E= -918.836438 a.u. ( 4.42 eV), f=0.0000, <S**2>= 0.000
Excited State 2: E= -918.836438 a.u. ( 4.42 eV), f=0.0000, <S**2>= 0.000
State-averaged NO generated using states 0~2.
MOKIT判断活性空间取(4e,3o)就够,看起来也确实是这样:
| orb index | type | occ |
|---|---|---|
| 20 | Virtual | 0.0006 |
| 19 | Virtual | 0.0061 |
| 18 | Virtual | 0.7899 |
| 17 | Occupied | 1.6046 |
| 16 | Occupied | 1.6046 |
| 15 | Occupied | 1.9964 |
| 14 | Occupied | 1.9964 |
| 13 | Occupied | 1.9990 |
NEVPT2计算结果:
1
2
3
4
5
6
7
8
9
10
11
SA-CASSCF(4e,3o) using program pyscf
$python Cl2_mokit_CIS_NO_SA-CAS.py >Cl2_mokit_CIS_NO_SA-CAS.out 2>&1
CASCI energies after SA-CASSCF(0 for ground state): E_ex/eV fosc
State 0, E = -918.99823059 a.u. <S**2> = 0.000
State 1, E = -918.83264461 a.u. <S**2> = 0.000 4.506 0.0011
State 2, E = -918.83264461 a.u. <S**2> = 0.000 4.506 0.0011
NEVPT2 energies: E_ex/eV
State 0, E = -919.44884920 a.u.
State 1, E = -919.30884747 a.u. 3.810
State 2, E = -919.30884747 a.u. 3.810
不过,如果要按照物理直觉的话,应该把3p全都拿进活性空间,也就是(10e,6o),那么可以指定:
1
2
3
4
5
6
7
8
9
%nproc=32
%mem=96GB
# NEVPT2(10,6)/cc-pvtz
mokit{Nstates=2}
0 1
Cl 0.000000 0.000000 0.994248
Cl 0.000000 0.000000 -0.994248
MOKIT会提示你(4e,3o)就够,但也会按照你指定的来扩展:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
Warning: SA-CAS(4e,3o) recommended, but you specify SA-CAS(10e,6o). Trying to fulfill...
...
OK, fulfilled.
...
SA-CASSCF(10e,6o) using program pyscf
$python Cl2_106_CIS_NO_SA-CAS.py >Cl2_106_CIS_NO_SA-CAS.out 2>&1
CASCI energies after SA-CASSCF(0 for ground state): E_ex/eV fosc
State 0, E = -919.01499298 a.u. <S**2> = 0.000
State 1, E = -918.85755861 a.u. <S**2> = 0.000 4.284 0.0008
State 2, E = -918.85755861 a.u. <S**2> = 0.000 4.284 0.0008
NEVPT2 energies: E_ex/eV
State 0, E = -919.42423482 a.u.
State 1, E = -919.27994084 a.u. 3.926
State 2, E = -919.27994084 a.u. 3.926
可以看到用自动判断的和手动指定的活性空间都算出了简并的$^1\Pi_u$态2。用(10e,6o)算的激发能是3.925 eV,比(4e,3o)的3.810 eV稍高。搜到exp:3.76 eV,这样看反而是(4e,3o)更接近实验值。不过此处没考虑Spin-Orbit Coupling,这个还会进一步降低$^1\Pi_u$的能量,外加NEVPT2也会略微高估激发能,(4e,3o)可能有些误差抵消的运气成分在。
我们还是取符合物理直觉的(10e,6o),不然不好统一活性空间。其激发能对应波长316 nm,在很蓝的位置。
解离势能面
做个scan看看势能面是不是跟笔者先前推测的一样会走一个CI。这里笔者把$^3\Pi_u$态也算了,检查下有没有MECP,产生别的解离通道。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
#!/bin/bash
# --- 参数设置区域 ---
R_eq=1.9885 # 起始键长
step=0.05 # 步长
n_points=35 # 总点数
base_name="Cl2"
list_file="scan_info.txt" # 记录列表的文件名
# ------------------
# 1. 初始化列表文件,写入表头
# 使用 printf 保证表头对齐
printf "%-10s %-15s %-20s\n" "Point_ID" "Distance(A)" "Filename" > "$list_file"
for ((i=1; i<=n_points; i++))
do
# 计算当前键长
current_L=$(awk -v r="$R_eq" -v s="$step" -v i="$i" 'BEGIN {printf "%.4f", r + (i-1)*s}')
# 生成文件名
file_id=$(printf "%03d" $i)
filename="${base_name}_${file_id}.gjf"
# 生成输入文件
cat > "$filename" <<EOF
%nproc=32
%mem=96GB
# NEVPT2(10,6)/cc-pvtz
mokit{Nstates=4,mixedspin}
0 1
Cl 0.0 0.0 0.0
Cl 0.0 0.0 $current_L
EOF
# 2. 将信息追加到列表文件中
# 这里的 %-10s 等是为了让文本列对齐,看起来更舒服
printf "%-10s %-15s %-20s\n" "$file_id" "$current_L" "$filename" >> "$list_file"
echo "已生成: $filename"
done
echo "----------------------------------------"
echo "所有文件已生成,键长列表已保存在: $list_file"
计算完成后,画个PES。绘图脚本太长就不贴了,读者可自行下载。PES长这样:
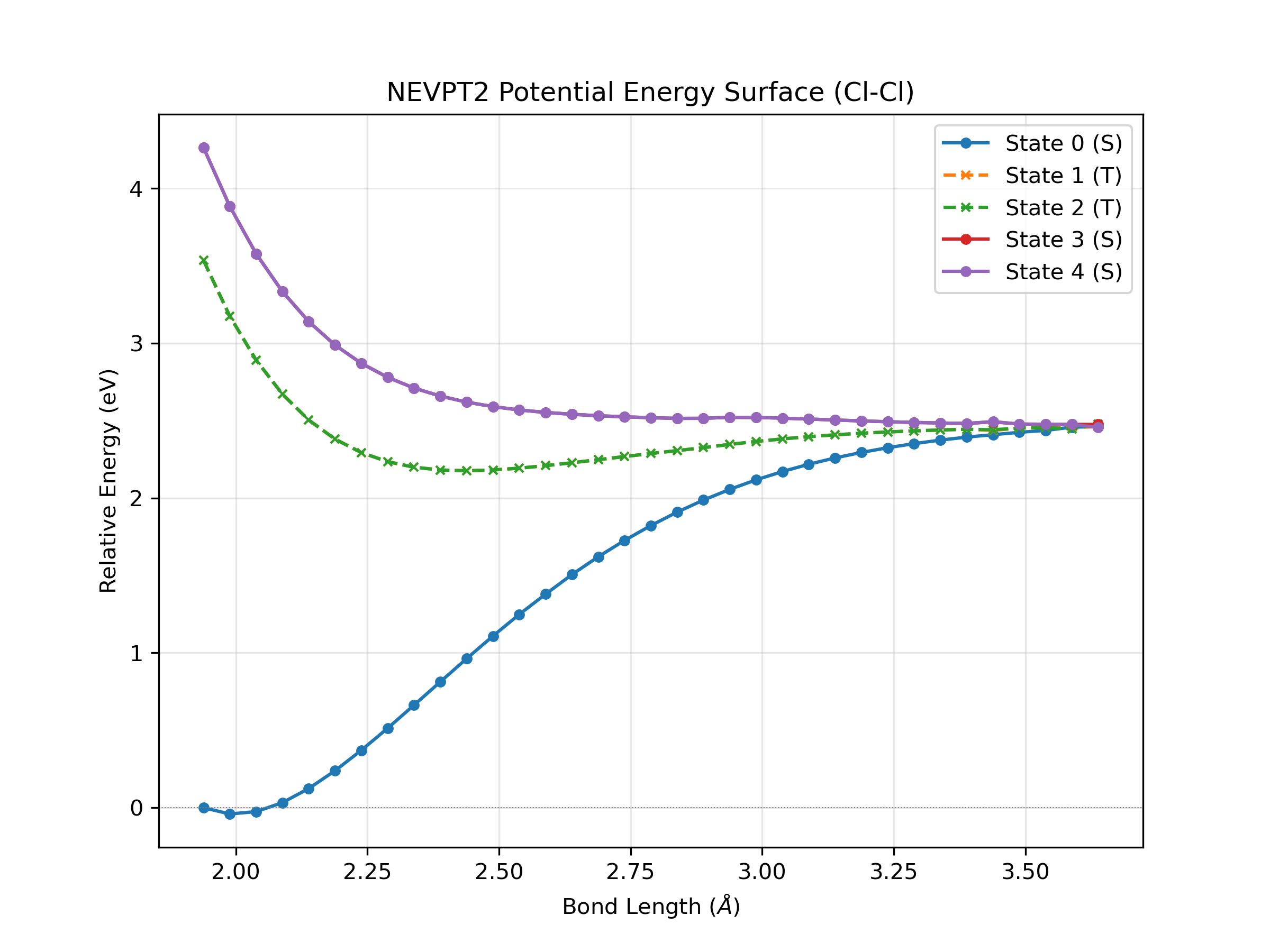
可以看到,三重态的能量始终比单重态没低多少。进一步计算SOC:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
%pal nprocs 16 end
%maxcore 6000
! CASSCF VeryTightSCF
%casscf
nel 10
norb 6
mult 1,3
nroots 3,2
maxiter 200
CI MaxIter 200 NGuessMat 1000 end
rel dosoc true PrintLevel 2 end
PTMethod SC_NEVPT2
end
%method
FrozenCore FC_NONE
end
%scf
Thresh 1e-12
Tcut 1e-14
CNVDamp False
end
先看一眼能量跟之pyscf计算值能不能对上,以确认两个程序是不是收敛到相同的解
1
2
3
4
5
6
7
8
9
10
11
-----------------------------
NEVPT2 TRANSITION ENERGIES
------------------------------
LOWEST ROOT (ROOT 0, MULT 1) = -919.424605715 Eh -25018.815 eV
STATE ROOT MULT DE/a.u. DE/eV DE/cm**-1
1: 0 3 0.118301 3.219 25964.2
2: 1 3 0.118301 3.219 25964.2
3: 1 1 0.144301 3.927 31670.5
4: 2 1 0.144301 3.927 31670.5
对比pyscf结果,可以看到基本一致
1
2
3
4
5
6
NEVPT2 energies: E_ex/eV
State 0, E = -919.42452578 a.u.
State 1, E = -919.30635102 a.u. 3.216
State 2, E = -919.30635102 a.u. 3.216
State 3, E = -919.28030095 a.u. 3.925
State 4, E = -919.28030095 a.u. 3.925
读取旋轨耦合,得S1/T1的socme值为106.47 cm-1。这个值是相当大的,说明这个ISC可以发生。从PES的形状上看,$^3\Pi_u$在2.35埃左右有个极小点,可能是因为同自旋的组合让$\sigma_{3p}^*$轨道削弱氯氯键的效应没那么强,允许氯气以一个弱键的形式存在。这是一个不利于解离的因素。
但也应当注意到这里的势垒非常低。笔者之前看过些研究MECI的文献,有一种意见是MECP点的能垒应当用E(MECP)-E(FC)而不是E(MECP)-E(S1),笔者的理解是FC点弛豫到S1极小点时还会带着动能,很容易走到势能面另一边相同势能的位置,所以这部分能垒不算数:
 J. Phys. Chem. Lett. 2020, 11, 7790−7797
J. Phys. Chem. Lett. 2020, 11, 7790−7797
参考文献说法,氯气这个能垒理论上非常容易跨过去,走到三态交叉点还是很容易的。同时,注意到S0/T1的socme为0,因此从T1直接跃迁回S0要靠HT效应,在数量级上不会比进入MECP的速率更高,最终还是会解离。
综上,氯气的光解大概两条路径:
- 直接从FC点沿$^1\Pi_u$势能面弛豫到MECP点
- 先通过ISC到$^3\Pi_u$,再跨过一个很浅的势垒走到MECP点
而这一切的前提都是,氯气需要被激发到$^1\Pi_u$态。
Mg的发射
接下来看看镁条的发射。Mg燃烧的时候温度非常高,可以通过热激发来到$^1P_1$态3,我们看看它的发射是什么样。Mg的活性空间应该是3s电子和3s+3p轨道,也就是(2e,4o)。从3s向任意一个3p激发,应当是三重简并的,所以SA-CAS平均基态和三个激发态:
1
2
3
4
5
6
7
8
%nproc=32
%mem=96GB
# NEVPT2(2,4)/aug-cc-pvtz
mokit{Nstates=3}
0 1
Mg 0.000000 0.000000 0.000000
得到激发能4.326 eV,换算波长285 nm,明显属于紫外发射。(搜到exp:4.436 eV,算是对的不错了)
1
2
3
4
5
6
7
8
9
10
11
12
13
SA-CASSCF(2e,4o) using program pyscf
$python Mg_CIS_NO_SA-CAS.py >Mg_CIS_NO_SA-CAS.out 2>&1
CASCI energies after SA-CASSCF(0 for ground state): E_ex/eV fosc
State 0, E = -199.61202158 a.u. <S**2> = 0.000
State 1, E = -199.46722928 a.u. <S**2> = 0.000 3.940 0.3976
State 2, E = -199.46722906 a.u. <S**2> = 0.000 3.940 0.3976
State 3, E = -199.46722880 a.u. <S**2> = 0.000 3.940 0.3976
NEVPT2 energies: E_ex/eV
State 0, E = -199.65822267 a.u.
State 1, E = -199.49923536 a.u. 4.326
State 2, E = -199.49923561 a.u. 4.326
State 3, E = -199.49923568 a.u. 4.326
注意到振子强度有0.3976,说明Mg在285 nm这个发射比较强。
但是这里有个新问题,Mg的发射峰在4.326 eV,Cl2的吸收峰在3.925 eV,这差了0.4 eV还是不小的,真的能匹配吗?笔者提出两个猜测:
- i. 氯气有个更高的激发态,能量恰好在4.3 eV附近
- ii. $^1\Pi_u$态的吸收带可以展到285 nm(4.3 eV)附近
这里先简单排除一下猜想1:
1
2
3
4
5
6
NEVPT2 energies: E_ex/eV
State 0, E = -919.42362289 a.u.
State 1, E = -919.27946423 a.u. 3.923
State 2, E = -919.27946422 a.u. 3.923
State 3, E = -919.17361624 a.u. 6.803
State 4, E = -919.17361624 a.u. 6.803
后面的态能量的有点太高了,所以大概是第二种原因,接下来验证一下。
吸收光谱
吸收峰已经有了,这里可以选择用高斯函数展出来一个模拟光谱,但我们最终的目的是找到引发氯气解离的光谱范围,FWHM随意取好像不太好。不过这个分子只有一个正则模式,而且前面freq=num已经算了频率值是561.4278 cm-1(exp:560 cm-1左右,对的不错)。于是这里可以玩点花的。首先基于力常数做个Wigner采样:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
import numpy as np
import os
# ================= 配置 =================
# 输入参数
freq_cm = 561.4 # 频率 (cm^-1)
mass_amu = 35.45 # 相对原子质量
r_eq = 1.9885 # 平衡键长
temp = 298.15 # 温度 (K)
n_samples = 200 # 采样数量
# 计算设置
nproc = 32
mem = "96GB"
route_section = "# NEVPT2(10,6)/cc-pvtz"
mokit_options = "mokit{Nstates=2}"
output_dir = "gjf_inputs" # 输出目录
# ================= 采样 =================
# 物理常数与 Wigner 采样参数
h_bar = 1.0545718e-34 # J*s
c = 2.99792458e10 # cm/s
amu_to_kg = 1.660539e-27 # kg
k_B = 1.380649e-23 # J/K
mu = (mass_amu * amu_to_kg) / 2.0
omega = freq_cm * 2 * np.pi * c
sigma_0k = np.sqrt(h_bar / (2 * mu * omega))
if temp == 0:
correction_factor = 1.0
else:
exponent = (h_bar * omega) / (2 * k_B * temp)
correction_factor = np.sqrt(1.0 / np.tanh(exponent))
sigma_r_meter = sigma_0k * correction_factor
sigma_r_ang = sigma_r_meter * 1e10
# Wigner采样并排序
deltas = np.random.normal(0, sigma_r_ang, n_samples)
bond_lengths = np.sort(r_eq + deltas)
print(f"--- Wigner 采样信息 ---")
print(f"温度: {temp} K")
print(f"频率: {freq_cm} cm^-1")
print(f"键长标准差 (Sigma): {sigma_r_ang:.4f} Å")
print(f"生成样本数: {n_samples}")
print("-" * 30)
# ================= 计算 =================
os.makedirs(output_dir, exist_ok=True)
for i, r in enumerate(bond_lengths):
# 格式化索引,200个文件建议用三位数字补零 (001, 002...) 方便排序
index = i + 1
filename = f"Cl2_point{index:03d}.gjf"
filepath = os.path.join(output_dir, filename)
# 构建文件内容
content = f"""%nproc={nproc}
%mem={mem}
{route_section}
{mokit_options}
0 1
Cl 0.000000 0.000000 0.000000
Cl 0.000000 0.000000 {r:.6f}
"""
# 写入文件
with open(filepath, 'w') as f:
f.write(content)
# 仅打印前几个和后几个作为进度提示,避免刷屏
if i < 3 or i >= n_samples - 3:
print(f"Generated: {filepath} (R = {r:.4f} Å)")
elif i == 3:
print("...")
print("-" * 30)
print(f"全部完成!文件已保存在 {output_dir}/ 目录下。")
随后运行计算并绘图。这里因为NEVPT2不输出振子强度,笔者用了CASCI的振子强度凑合。绘图脚本很长,不贴在这里了,读者可自行下载。
1
python spectrum.py
我们看最终的光谱:
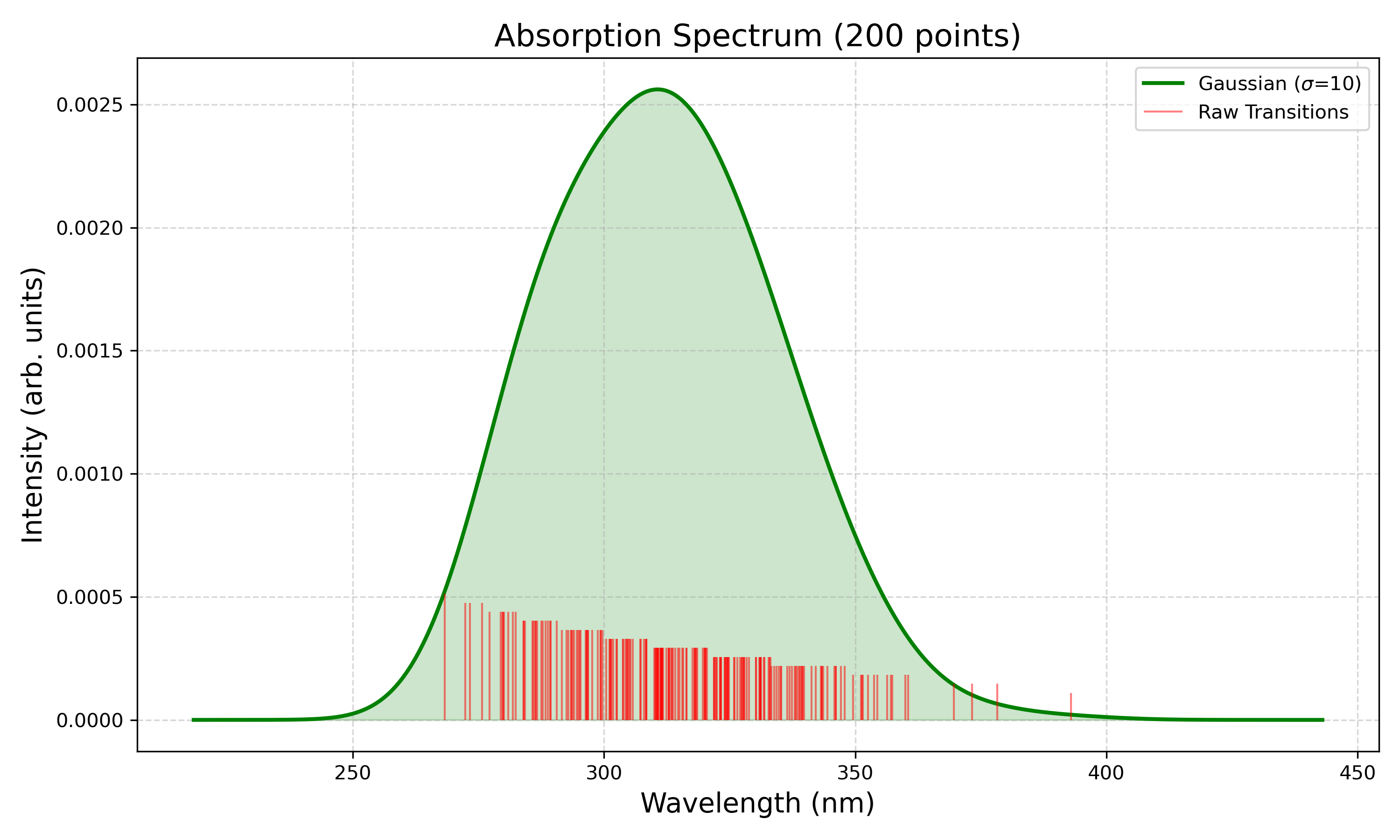
从光谱形状上看,$^1\Pi_u$态从250 nm开始有吸收,到400 nm为止,主要的吸收带大概在275-340 nm。搜了个实验光谱(下图紫线)。注意到模拟的光谱250 nm处吸收的提升速度略快于实验光谱,这可能是NEVPT2轻微高估激发能引起的。除此之外,特征非常接近。 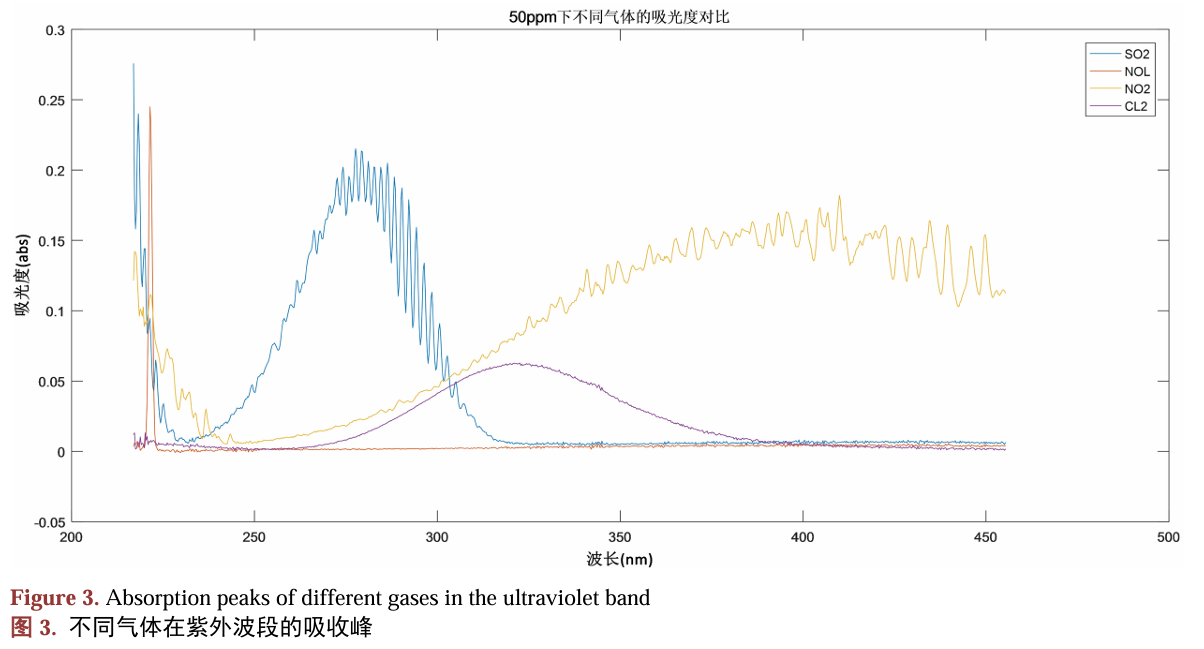 阎杰, 夏赛, 程鹏, 陈民旺. 基于差分吸收光谱技术的氯气在线监测系统研制[J]. 应用物理, 2023, 13(11): 421-429.
阎杰, 夏赛, 程鹏, 陈民旺. 基于差分吸收光谱技术的氯气在线监测系统研制[J]. 应用物理, 2023, 13(11): 421-429.
结论
结合前面的信息,可以总结如下:
- 镁的强发射在285 nm,恰好落在此范围内,是引发此反应很好的选择。
- 自然光在此范围的辐射强度比可见区弱了5、6倍,而且由于普通玻璃会吸收紫外线,能够透过窗户到达反应容器的会更少,笔者觉得想像镁一样引发这个反应到爆炸的程度可能不太容易,估计那时候可见光都开始晃眼了。
- LED电灯的发射峰在450 nm,虽然能量高于氯气的键能,但氯气在400以上吸收只能靠$^3\Pi_u$的禁阻吸收(氯气呈淡黄绿色的原因),吸收截面非常非常小,而电灯在400以下也基本无发射,想引发这个反应是非常困难的。
ASTM G-173是一个比较常用的太阳光谱实验测试数据,Gemini称这是“标准太阳光”。笔者在NREL官网找到了这个数据:Reference Air Mass 1.5 Spectra ↩︎
$^1\Pi_u$ 是一个分子谱项记号,左上角的$^1$指单重态;$\Pi$ 是旋转对称性记号,指角动量投影$\Lambda=1$;右下角的$u$是反演对称性记号,$u$是奇宇称。来历:氯气的第一激发态是H→L的跃迁,根据分子轨道理论,氯气的FMO分子轨道能量顺序是 $ \sigma_{3s} < \sigma_{3s}^* < \sigma_{3p} < \pi_{3p} < \pi_{3p}^* < \sigma_{3p}^* $。填充14个价电子后,HOMO轨道是二重简并的$\pi_{3p}^* $(这也是$^1\Pi_u$态二重简并的原因),而LUMO轨道是$\sigma_{3p}^* $。$\sigma$轨道的轨道角动量是0,$\pi$轨道的轨道角动量是±1,最终$\Lambda=1$,为$\Pi$态。$\sigma$轨道是偶宇称,$\pi$轨道是奇宇称,所以这个态也是奇宇称。 ↩︎
$^1P_1$是一个原子谱项记号,左上角的$^1$依然指单重态($S=0$);$P$是指轨道角动量$L=1$;右下角则是总角动量$J$,$J=L+S=1$。 ↩︎